TURBOMOLE & phonopy calculation#
Supported features#
The riper module of TURBOMOLE can be used to study periodic structures.
The TURBOMOLE interface reads the unit cell from TURBOMOLE input files.
Lattice vectors and the name of the coordinate file are read from the control file.
Atomic symbols and Cartesian coordinates are read from the coordinate file.
How to run#
The workflow for a TURBOMOLE-Phonopy calculation is outlined here using the
Si example found in example/Si-TURBOMOLE.
In this example, the TURBOMOLE input files are control and coord.
This is the default for the TURBOMOLE interface and therefore the -c control
parameter is not needed.
Create supercells with --turbomole option:
% phonopy-init --turbomole --dim="3 3 3" -d
In this example, the Si crystal structure is defined with the conventional unit cell of eight atoms and 3x3x3 supercells are created. For every supercell, the interface creates a subdirectory with
controlandcoordfiles. Files insupercellcontain the perfect supercell. The files insupercell-xxx(xxxare numbers) contain the supercells with displacements. Filephonopy_disp.yamlis also generated, containing information about the supercell and the displacements.In the case of the Si example, subdirectory
supercell-001will be created.Complete TURBOMOLE inputs need to be prepared manually in the subdirectories.
Note that supercells with displacements must not be relaxed, because the atomic forces induced by a small atomic displacement are what we need for phonon calculation. To get accurate forces, $scfconv should be at least 10. Phonopy includes this data group automatically in the control file. You also need to choose a k-point mesh for the force calculations. TURBOMOLE data group $riper may need to be adjusted to improve SCF convergence (see example files in subdirectory
supercell-001for further details)Then, TURBOMOLE supercell calculations are executed to obtain forces on atoms, e.g., as follows:
% riper > supercell-001.out
To create
FORCE_SETSfile required by Phonopy, the following command is executed:% phonopy-init --turbomole -f supercell-*
Here
supercell-*directories contain the TURBOMOLE output files from the force calculations (only the filegradientis required). To run this command,phonopy_disp.yamlhas to be located in the current directory because the information on atomic displacements stored in this file are used to generateFORCE_SETS. See some more detail at TURBOMOLE interface.Now, Phonopy post-prcessing commands can be run.
FORCE_SETSis automatically read in. Note that here PRIMITIVE_AXES is defined in band.conf to create the phonon dispersions for the primitive cell.Create phonon dispersion plot with:
% phonopy -p -s band.conf _ _ __ | |__ ___ _ __ ___ _ __ _ _ | '_ \| '_ \ / _ \| '_ \ / _ \ | '_ \| | | | | |_) | | | | (_) | | | | (_) || |_) | |_| | | .__/|_| |_|\___/|_| |_|\___(_) .__/ \__, | |_| |_| |___/ 2.1.1 Python version 3.7.1 Spglib version 1.12.1 Calculator interface: turbomole Crystal structure was read from "control". Band structure mode Settings: Supercell: [3 3 3] Primitive matrix: [0. 0.5 0.5] [0.5 0. 0.5] [0.5 0.5 0. ] Spacegroup: Fd-3m (227) Use -v option to watch primitive cell, unit cell, and supercell structures. Forces and displacements are read from "FORCE_SETS". Computing force constants... Max drift of force constants: -0.000000 (xx) -0.000000 (xx) Reciprocal space paths in reduced coordinates: [ 0.000 0.000 0.000] --> [ 0.500 0.000 0.500] [ 0.500 0.000 0.500] --> [ 0.500 0.250 0.750] [ 0.500 0.250 0.750] --> [ 0.375 0.375 0.750] [ 0.375 0.375 0.750] --> [ 0.000 0.000 0.000] [ 0.000 0.000 0.000] --> [ 0.500 0.500 0.500] [ 0.500 0.500 0.500] --> [ 0.625 0.250 0.625] [ 0.625 0.250 0.625] --> [ 0.500 0.250 0.750] [ 0.500 0.250 0.750] --> [ 0.500 0.500 0.500] [ 0.500 0.500 0.500] --> [ 0.375 0.375 0.750] _ ___ _ __ __| | / _ \ '_ \ / _` | | __/ | | | (_| | \___|_| |_|\__,_|
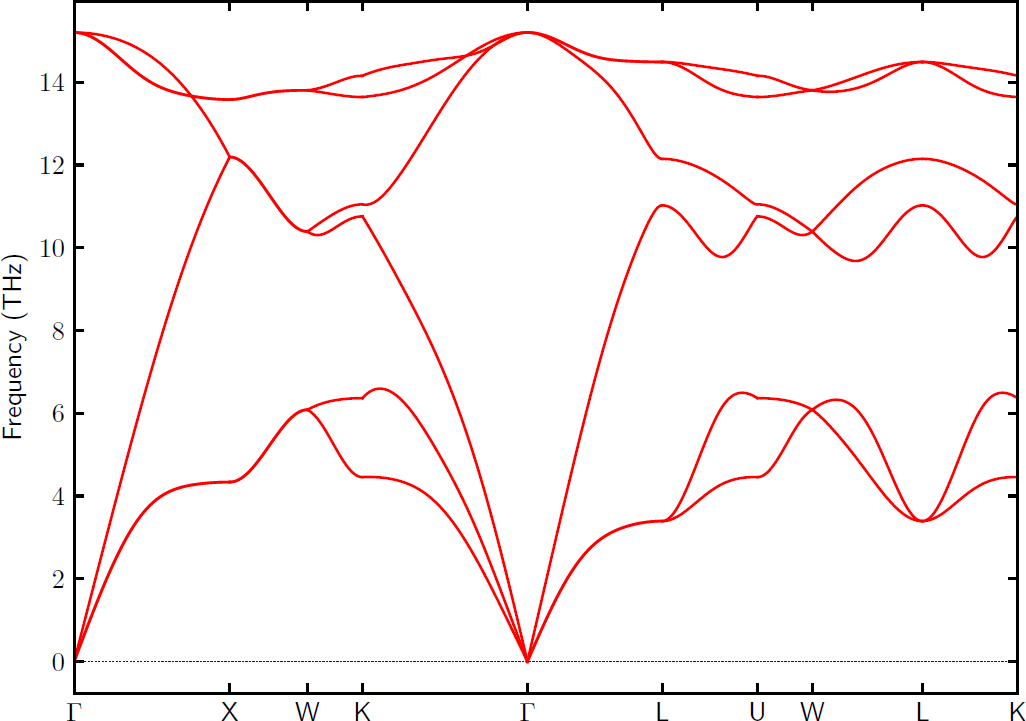
For further settings and command options, see the general Phonopy documentation Setting tags and Command options, respectively, and for examples, see Examples.