Calculation of mode Grüneisen parameters#
How to run#
It is necessary to run three phonon calculations. One is calculated at the equilibrium volume and the remaining two are calculated at the slightly larger volume and smaller volume than the equilibrium volume. The unit cells at these volumes have to be fully relaxed under the constraint of each volume.
Let files named POSCAR-unitcell, FORCE_SETS (or FORCE_CONSTANTS with
--readfc option), and optionally BORN stored in three different directories
named, e.g., equiv, plus, and minus.
The calculated results are written into the file gruneisen.yaml.
In the example directory, an example of silicon (Si-gruneisen) is prepared. A
calculation along paths in reciprocal space can be made by
% phonopy-gruneisen orig plus minus --dim="2 2 2" --pa="0 1/2 1/2 1/2 0 1/2 1/2 1/2 0" --band="1/2 1/4 3/4 0 0 0 1/2 1/2 1/2 1/2 0.0 1/2" -p -c POSCAR-unitcell
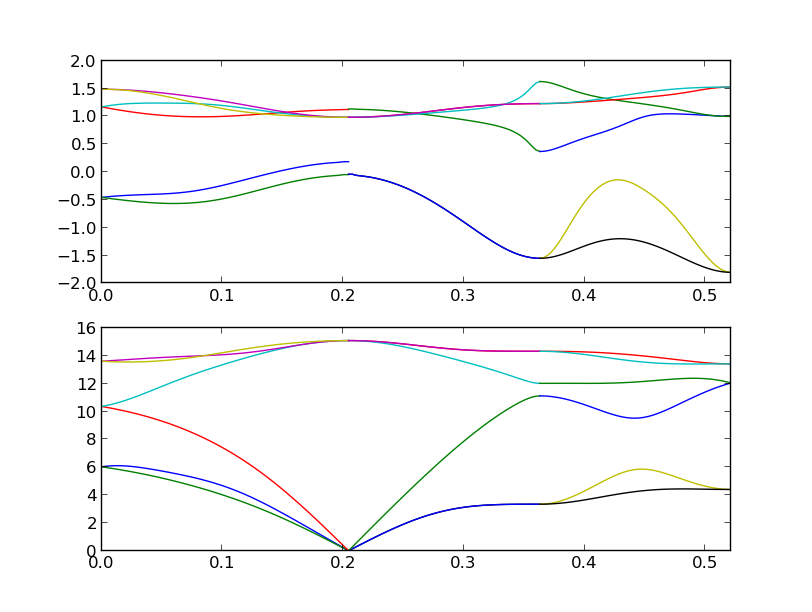
In this calculation, neighboring q-points in each band segment are connected
considering their phonon symmetry to treat band crossing correctly. Therefore
the phonon frequencies may not be ordered in gruneisen.yaml. In the plot (-p
option), the colors of phonon bands correspond to those of mode Grüneinen
parameters.
A calculation on a reciprocal mesh is made by
% phonopy-gruneisen orig plus minus --dim="2 2 2" --pa="0 1/2 1/2 1/2 0 1/2 1/2 1/2 0" --mesh="20 20 20" -p -c POSCAR-unitcell --color="RB"
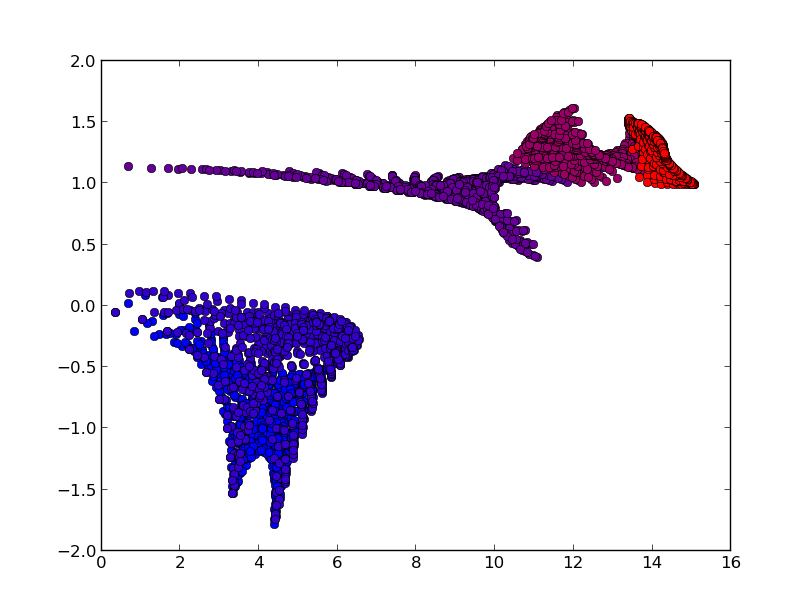
In the plot (-p option), the colors of mode Grüneinen parameters are set for
band indices with ascending order of phonon frequencies.
Mode Grüneinen parameter may diverge around \(\Gamma\)-point. In the above
example for band paths, mode Grüneinen parameters are calculated at
\(\Gamma\)-point, but phonopy-gruneisen script avoids showing the values
on the plot. Instead the values at the neighboring q-points of
\(\Gamma\)-point are used for the plot.
Force calculator interfaces#
Options of force calculator interfaces such as --abinit, --qe, … can be
specified for corresponding calculators and the crystal structure file format
should be different from that of the VASP format. An Abinit example is as
follows:
% phonopy-gruneisen orig plus minus --abinit --dim="2 2 2" --pa="0 1/2 1/2 1/2 0 1/2 1/2 1/2 0" --band="1/2 1/4 3/4 0 0 0 1/2 1/2 1/2 1/2 0.0 1/2" -p -c Si.in
Command options#
If one of the force calculator options is specified, the interface mode is
changed to it. The unit conversion factor to THz is appropriately selected and
its crystal structure file format is accepted. If none of them is specified, as
the VASP interface mode is invoked as the default interface. The --factor
option was deprecated at v2.44.
The following command options can be used for all interface modes. They work
similarly to those for phonopy script.
--dim--mp,--mesh--band--pa,--primitive_axis--readfc--band_points--nac--nomeshsym-p-c-s,--save-o
Currently --pa="auto" works but --band="auto" doesn’t. The --color option
(RB, RG, RGB) is used to gradually change the marker colors with respect
to band indices. For the mesh-sampling plot, a few more options to control
matplotlib parameters are prepared.
Method to calculate mode Grüneisen parameters#
Mode Grüneisen parameter \(\gamma(\mathbf{q}\nu)\) at the wave vector \(\mathbf{q}\) and band index \(\nu\) is given by
where \(V\) is the volume, \(\omega(\mathbf{q}\nu)\) is the phonon frequency, \(D(\mathbf{q})\) is the dynamical matrix, and \(\mathbf{e}(\mathbf{q}\nu)\) is the eigenvector. This is approximated by the finite difference method:
The phonopy-gruneisen script requires three phonon calculations at
corresponding three volume points. One is for eigenvectors at the equilibrium
volume (\(V\)) and the remaining two are for \(\Delta D(\mathbf{q})\)
with slightly larger and smaller volumes than \(V\).