Setting tags#
Configuration file#
Most of the setting tags have respective command-line options (Command options). When both of equivalent command-line option and setting tag are set simultaneously, the command-line option supersedes the setting tag. The configuration file is recommended to place at the first position for the mixed use of setting tags and command-line options, i.e.,
% phonopy --config setting.conf [OPTIONS]
For specifying real and reciprocal points, fractional values (e.g. 1/3) are
accepted. However fractional values must not have space among characters (e.g.
1 / 3) are not allowed.
Band structure tags#
BAND#
BAND gives sampling band paths. The reciprocal points are specified in reduced
coordinates. The given points are connected for defining band paths. When comma
, is inserted between the points, the paths are disconnected.
An example of three paths, (0,0,0) to (1/2,0,1/2), (1/2,1/2,1) to (0,0,0), and (0,0,0) to (1/2,1/2,1/2), with 101 sampling points of each path are as follows:
BAND = 0 0 0 1/2 0 1/2, 1/2 1/2 1 0 0 0 1/2 1/2 1/2
BAND_POINTS = 101
BAND_POINTS#
BAND_POINTS gives the number of sampling points including the path ends. The
default value is BAND_POINTS = 51.
BAND_LABELS#
Labels specified are depicted in band structure plot at the points of band
segments. The number of labels has to correspond to the number of band paths
specified by BAND plus one. When LaTeX math style expression such as
\(\Gamma\) (\Gamma) is expected, it is probably necessary to place it
between two $ characters.
BAND = 1/2 0 1/2 0 0 0 1/2 1/2 1/2
BAND_LABELS = X $\Gamma$ L
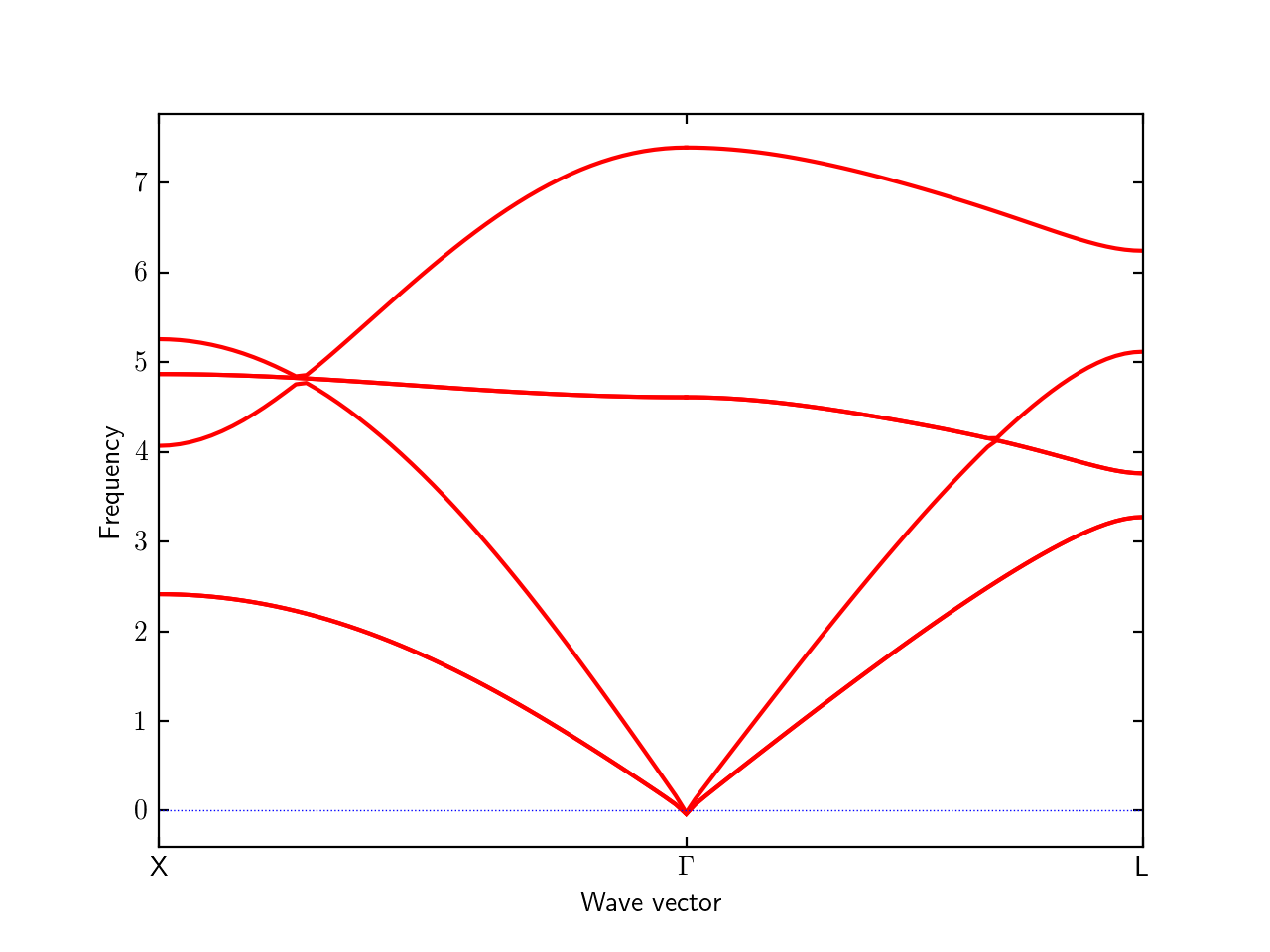
The colors of curves are automatically determined by matplotlib. The same color in a band segment shows the same kind of band. Between different band segments, the correspondence of colors doesn’t mean anything.
BAND_CONNECTION#
With this option, band connections are estimated from eigenvectors and band
structure is drawn considering band crossings. In sensitive cases, to obtain
better band connections, it requires to increase number of points calculated in
band segments by the BAND_POINTS tag.
BAND = 1/2 0 1/2 0 0 0 1/2 1/2 1/2
BAND_POINTS = 101
BAND_CONNECTION = .TRUE.
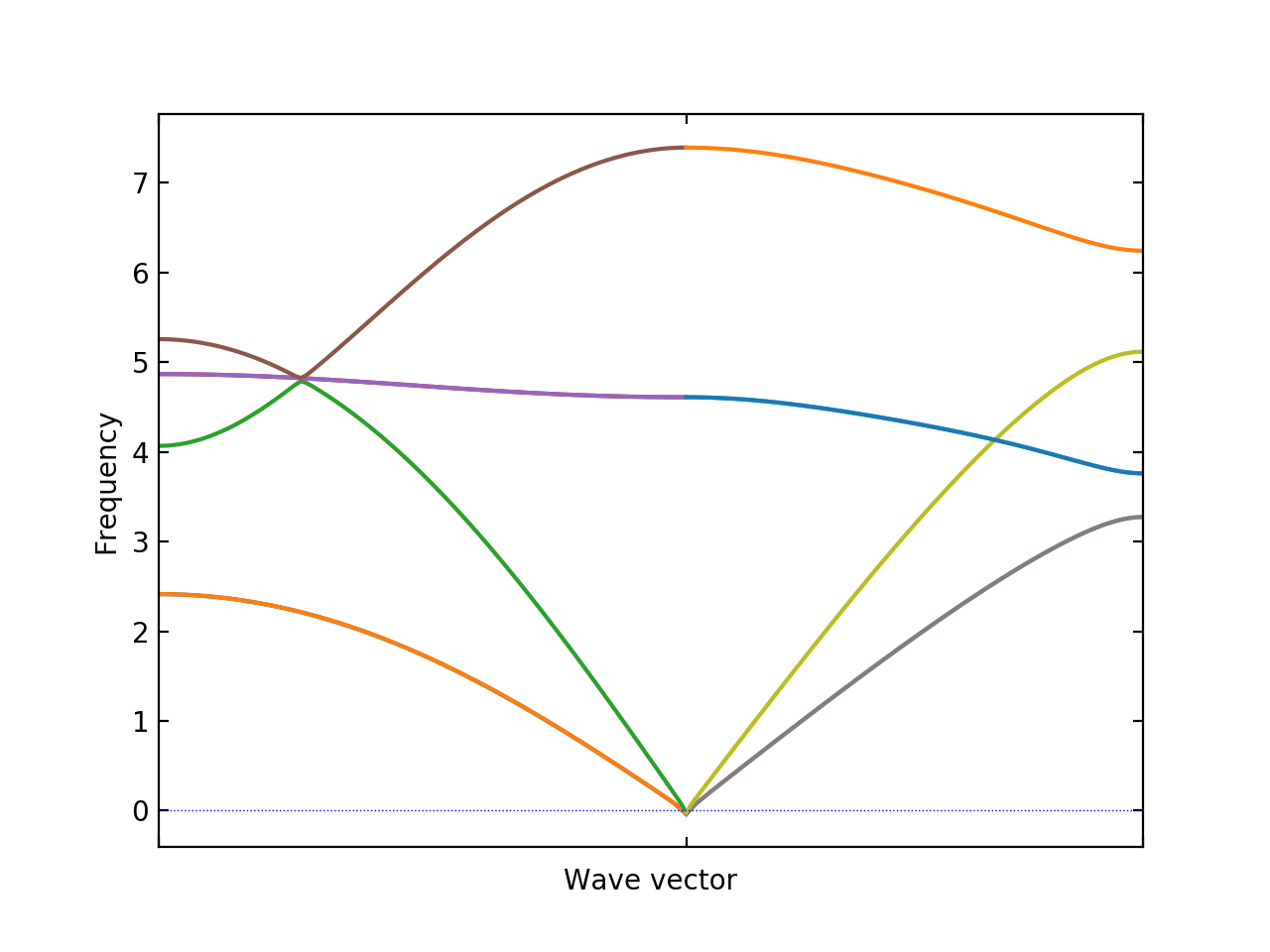
BAND_CONST_INTERVAL#
When BAND_CONST_INTERVAL = .TRUE., the number of q-points sampled in each band
path segment is determined by the longest path segment, which uses the number
specified by BAND_POINTS. This ensures similar distances between points across
all segments. Consequently, the number of q-points in each band path segment is
equal to or less than the value of BAND_POINTS. The default value is
.FALSE..
BAND = AUTO
BAND_POINTS = 101
BAND_CONST_INTERVAL = .TRUE.
Thermal displacements#
TDISP#
Mean square displacements projected to Cartesian axes as a function of
temperature are calculated from the number of phonon excitations. Phonon
frequencies in THz, which is the default setting of phonopy, are used to obtain
the mean square displacements, therefore physical units have to be set properly
for it (see Interfaces to calculators.) The result is given in
\(\text{Angstrom}^2\) and writen into thermal_displacements.yaml. See the
detail of the method, Thermal displacement. These tags must be used with
Mesh sampling tags
Optionally, FMIN tag (--fmin option) with a small value is recommened to be
set when q-points at \(\Gamma\) point or near \(\Gamma\) point (e.g.
using very dense sampling mesh) are sampled to avoid divergence. FMAX tag
(--fmax option) can be used to specify an upper bound of phonon frequencies
where the phonons are considered in the summation. The projection is applied
along arbitrary direction using PROJECTION_DIRECTION tag
(PROJECTION_DIRECTION).
TMAX, TMIN, TSTEP tags are used to control temperature points at which the
thermal displacements are calculated.
mesh.yaml or mesh.hdf5 is not written out from phonopy-1.11.14.
TDISP = .TRUE.
PROJECTION_DIRECTION = 1 1 0
TDISPMAT#
Mean square displacement matrices are calculated. The definition is shown at
Thermal displacement. Phonon frequencies in THz, which is the default
setting of phonopy, are used to obtain the mean square displacement matrices,
therefore physical units have to be set properly for it (see
Interfaces to calculators.) The result is given in \(\text{Angstrom}^2\)
and written into thermal_displacement_matrices.yaml where six matrix elements
are given in the order of xx, yy, zz, yz, xz, xy. In this yaml file,
displacement_matrices and displacement_matrices_cif correspond to
\(\mathrm{U}_\text{cart}\) and \(\mathrm{U}_\text{cif}\) defined at
Mean square displacement matrix, respectively.
Optionally, FMIN tag (--fmin option) with a small value is recommended to be
set when q-points at \(\Gamma\) point or near \(\Gamma\) point (e.g.
using very dense sampling mesh) are sampled to avoid divergence. FMAX tag
(--fmax option) can be used to specify an upper bound of phonon frequencies
where the phonons are considered in the summation.
The 3x3 matrix restricts distribution of each atom around the equilibrium position to be ellipsoid. But the distribution is not necessarily to be so.
TMAX, TMIN, TSTEP tags are used to control temperature points at which the
thermal displacement matrices are calculated.
mesh.yaml or mesh.hdf5 is not written out from phonopy-1.11.14.
TDISPMAT = .TRUE.
TDISPMAT_CIF#
This tag specifies a temperature (K) at which thermal displacement is calculated
and the mean square displacement matrix is written to the cif file
tdispmat.cif with the dictionary item aniso_U. Phonon frequencies in THz,
which is the default setting of phonopy, are used to obtain the mean square
displacement matrices, therefore physical units have to be set properly for it
(see Interfaces to calculators.) The result is given in
\(\textrm{Angstrom}^2\).
mesh.yaml or mesh.hdf5 is not written out from phonopy-1.11.14.
TDISPMAT_CIF = 1273.0
Specific q-points#
QPOINTS#
When q-points are supplied, those phonons are calculated. Q-points are specified successive values separated by spaces and collected by every three values as vectors in reciprocal reduced coordinates.
QPOINTS = 0 0 0 1/2 1/2 1/2 1/2 0 1/2
With QPOINTS = .TRUE., q-points are read from QPOITNS file (see the file
format at QPOINTS) in current directory phonons at the
q-points are calculated.
QPOINTS = .TRUE.
WRITEDM#
WRITEDM = .TRUE.
Dynamical matrices \(D\) are written into qpoints.yaml in the following
\(6N\times3N\) format, where N is the number of atoms in the primitive
cell.
The physical unit of dynamical matrix is
[unit of force] / ([unit of displacement] * [unit of mass]), i.e., square of
the unit of phonon frequency before multiplying the unit conversion factor (see
FREQUENCY_CONVERSION_FACTOR).
and \(D_{jj'}\) is
where j and j’ are the atomic indices in the primitive cell. The phonon
frequencies may be recovered from qpoints.yaml by writing a simple python
script. For example, qpoints.yaml is obtained for NaCl at
\(q=(0, 0.5, 0.5)\) by
phonopy --qpoints="0 1/2 1/2" --writedm
and the dynamical matrix may be used as
import yaml
import numpy as np
data = yaml.load(open("qpoints.yaml"))
dynmat = []
dynmat_data = data['phonon'][0]['dynamical_matrix']
for row in dynmat_data:
vals = np.reshape(row, (-1, 2))
dynmat.append(vals[:, 0] + vals[:, 1] * 1j)
dynmat = np.array(dynmat)
eigvals, eigvecs, = np.linalg.eigh(dynmat)
frequencies = np.sqrt(np.abs(eigvals.real)) * np.sign(eigvals.real)
conversion_factor_to_THz = 15.633302
print(frequencies * conversion_factor_to_THz)
Non-analytical term correction#
NAC#
Non-analytical term correction is applied to dynamical matrix. BORN file has
to be prepared in the current directory. See BORN (optional) and
Non-analytical term correction. The default method is
NAC_METHOD = GONZE after v1.13.0.
NAC = .TRUE.
NAC_METHOD#
The method of non-analytical term correction is chosen by this tag between two,
NAC_METHOD = GONZE (Correction by dipole-dipole interaction) and NAC_METHOD = WANG
(Interpolation scheme at general q-points with non-analytical term correction), and the default is the former after v1.13.0.
Q_DIRECTION#
This tag is used to activate non-analytical term correction (NAC) at
\(\mathbf{q}\rightarrow\mathbf{0}\), i.e. practically \(\Gamma\)-point,
because NAC is direction dependent. With this tag, \(\mathbf{q}\) is
specified in the fractional coordinates of the reciprocal basis vectors. Only
the direction has the meaning. Therefore Q_DIRECTION = 1 1 1 and
Q_DIRECTION = 2 2 2 give the same result. This tag is valid for QPOINTS,
IRREPS, and MODULATION tags.
Away from \(\Gamma\)-point, this setting is ignored and the specified q-point is used as the q-direction.
QPOINTS = 0 0 0 NAC = .TRUE.
Q_DIRECTION = 1 0 0
Group velocity#
GROUP_VELOCITY#
Group velocities at q-points are calculated by using this tag. The group velocities are written into a yaml file corresponding to the run mode in Cartesian coordinates. The physical unit depends on physical units of input files and frequency conversion factor. Usually the phonon frequency is given in THz. Therefore, the physical unit of the group velocity written in the output files is [unit-of-distance.THz]. The distance units for different force calculators are listed at Physical unit system for calculator. For example, VASP [Angstrom.THz], and QE [au.THz].
GROUP_VELOCITY = .TRUE.
Technical details are shown at Group velocity.
GV_DELTA_Q#
The reciprocal distance used for the finite difference method is specified. Unless this tag is set, the analytical derivative of the dynamical matrix is used (including the Gonze-Lee non-analytical term correction, for which the analytical dipole-dipole derivative is now available). Set this tag to fall back to the finite-difference path with the given step.
In earlier versions (v4.0.1 and earlier) the finite-difference path was
used implicitly with GV_DELTA_Q = 1e-5 whenever the Gonze-Lee
non-analytical term correction was active. Set GV_DELTA_Q = 1e-5
explicitly to reproduce that behavior.
GV_DELTA_Q = 0.01
Symmetry#
SYMMETRY_TOLERANCE#
This is used to set geometric tolerance to find symmetry of crystal structure.
The default value is 1e-5. In general, it is not a good idea to loosen the
tolerance. It is recommended to symmetrize crystal structure before starting
phonon calculation, e.g., using --symmetry option.
SYMMETRY_TOLERANCE = 1e-3
SYMMETRY#
P1 symmetry is enforced to the input unit cell by setting SYMMETRY = .FALSE.
MESH_SYMMETRY#
Symmetry search on the reciprocal sampling mesh is disabled by setting
MESH_SYMMETRY = .FALSE.. In some case such as hexagonal systems or primitive
cells of cubic systems having F and I-centrings, the results with and without
mesh symmetry give slightly different values for those properties that can
employ mesh symmetry. This happens when the uniform sampling mesh made along
basis vectors doesn’t have the same crystallographic point group as the crystal
itself. This symmetry breaking may be also seen by the fact that weight
written in mesh.yaml can be different from possible order of product group of
site-symmetry group and time reversal symmetry. Generally the difference becomes
smaller when increasing the sampling mesh numbers.
FC_SYMMETRY#
Note
Starting with version 1.12.3, this tag was changed to a boolean (.TRUE. or
.FALSE.), whereas it previously accepted an argument.
This tag is used to symmetrize force constants by translational symmetry and
permutation symmetry with .TRUE. or .FALSE..
FC_SYMMETRY = .TRUE.
From the translation invariance condition,
where i and j are the atom indices, and \(\alpha\) and \(\beta\) are the Cartesian indices for atoms i and j, respectively. When this condition is broken, the sum gives non-zero value. This value is subtracted from the diagonal blocks. Force constants are symmetric in each pair as
Mind that the other symmetries of force constants, i.e., the symmetry from
crystal symmetry or rotational symmetry, are broken to use FC_SYMMETRY.
Force constants#
FORCE_CONSTANTS#
FORCE_CONSTANTS = READ
There are three values to be set, which are READ and WRITE, and .FALSE..
The default is .FALSE.. When FORCE_CONSTANTS = READ, force constants are
read from FORCE_CONSTANTS file. With FORCE_CONSTANTS = WRITE, force
constants calculated from FORCE_SETS are written to FORCE_CONSTANTS file.
The file format of FORCE_CONSTANTS is shown
here.
FULL_FORCE_CONSTANTS#
FULL_FORCE_CONSTANTS = .TRUE. is used to compute full supercell constants
matrix. This may be used to enforce space group symmetry to existing force
constants.
The default setting is .FALSE.. By .TRUE. or .FALSE., the array
shape becomes (n_patom, n_satom, 3, 3) or (n_satom, n_satom, 3, 3),
respectively. The detail is found at FORCE_CONSTANTS and force_constants.hdf5.
READ_FORCE_CONSTANTS#
READ_FORCE_CONSTANTS = .TRUE. is equivalent to FORCE_CONSTANTS = READ.
WRITE_FORCE_CONSTANTS#
WRITE_FORCE_CONSTANTS = .TRUE. is equivalent to FORCE_CONSTANTS = WRITE.
FC_CALCULATOR#
External force constants calculator can be used using this tag. Currently
symfc and ALM are supported. The phonopy’s default force constants
calculator is based on finite difference method, for which atomic displacements
are made systematically. The following is the list of the force constants
calculator currently possible to be invoked from phonopy.
FC_CALCULATOR_OPTIONS#
To be written.
SYMFC#
New in v2.25 Symfc (symfc/symfc) is based on fitting approach and any displacements set of atoms in supercell can be handled. For example, random displacements generated by RANDOM_DISPLACEMENTS can be used to compute force constants. To use symfc, its python module has to be installed via conda-forge or pip.
When symfc is used, please cite the manuscript: A. Seko and A. Togo, Phys. Rev. B, 110, 214302 (2024) [doi]
FC_CALCULATOR = SYMFC
ALM#
New in v2.3 ALM (ttadano/ALM) is based on fitting approach and any displacements set of atoms in supercell can be handled. For example, random displacements generated by RANDOM_DISPLACEMENTS can be used to compute force constants. To use ALM, its python module has to be installed via conda-forge or building it. The installation instruction is found here.
When ALM is used, please cite the paper: T. Tadano and S. Tsuneyuki, J. Phys. Soc. Jpn. 87, 041015 (2018).
FC_CALCULATOR = ALM
Create animation file#
ANIME_TYPE#
ANIME_TYPE = JMOL
There are V_SIM, ARC, XYZ, JMOL, and POSCAR settings. Those may be
viewed by v_sim, gdis, jmol (animation), jmol (vibration), respectively.
For POSCAR, a set of POSCAR format structure files corresponding to
respective animation images are created such as APOSCAR-000,
APOSCAR-001,….
There are several parameters to be set in the ANIME tag.
ANIME#
The format of ANIME tag was modified after ver. 0.9.3.3.
For v_sim#
ANIME = 0.5 0.5 0
The values are the q-point to be calculated. An animation file of
anime.ascii is generated.
For the other animation formats#
Phonon is only calculated at \(\Gamma\) point. So q-point is not necessary to be set.
anime.arc, anime.xyz, anime.xyz_jmol, or APOSCAR-* are generated
according to the ANIME_TYPE setting.
ANIME = 4 5 20 0.5 0.5 0
The values are as follows from left:
Band index given by ascending order in phonon frequency.
Magnitude to be multiplied. In the harmonic phonon calculation, there is no amplitude information obtained directly. The relative amplitude among atoms in primitive cell can be obtained from eigenvectors with the constraint of the norm or the eigenvectors equals one, i.e., number of atoms in the primitive is large, the displacements become small. Therefore this has to be adjusted to make the animation good looking.
Number of images in one phonon period.
(4-6) Shift of atomic points in reduced coordinate in real space. These values can be omitted and the default values are
0 0 0.
For anime.xyz_jmol, the first and third values are not used, however dummy
values, e.g. 0, are required.
Create modulated structure#
MODULATION#
The MODULATION tag is used to create a crystal structure with displacements
along normal modes at q-point in the specified supercell dimension.
Atomic displacement of the j-th atom is created from the real part of the eigenvectors with amplitudes and phase factors as
where \(A\) is the amplitude, \(\phi\) is the phase, \(N_\mathrm{a}\) is the number of atoms in the supercell specified in this tag and \(m_j\) is the mass of the j-th atom, \(\mathbf{q}\) is the q-point specified, \(\mathbf{r}_{jl}\) is the position of the j-th atom in the l-th unit cell, and \(\mathbf{e}_j\) is the j-th atom part of eigenvector. Convention of eigenvector or dynamical matrix employed in phonopy is shown in Dynamical matrix.
If several modes are specified as shown in the example above, they are
overlapped on the structure. The output filenames are MPOSCAR and
MPOSCAR-<number>. Each modulated structure of a normal mode is written in
MPOSCAR-<number> where the numbers correspond to the order of specified sets
of modulations. MPOSCAR is the structure where all the modulations are summed.
MPOSCAR-orig is the structure without containing modulation, but the dimension
is the one that is specified. Some information is written into
modulation.yaml.
Usage#
The first three (nine) values correspond to supercell dimension (supercell
matrix) like the DIM tag. The following values are used to
describe how the atoms are modulated. Multiple sets of modulations can be
specified by separating by comma ,. In each set, the first three values give a
Q-point in the reduced coordinates in reciprocal space. Then the next three
values are the band index from the bottom with ascending order, amplitude, and
phase factor in degrees. The phase factor is optional. If it is not specified, 0
is used.
Before multiplying user specified phase factor, the phase of the modulation
vector is adjusted as the largest absolute value,
\(\left|\mathbf{e}_j\right|/\sqrt{m_j}\), of element of 3N dimensional
modulation vector to be real. The complex modulation vector is shown in
modulation.yaml.
MODULATION = 3 3 1, 1/3 1/3 0 1 2, 1/3 1/3 0 2 3.5
MODULATION = 3 3 1, 1/3 1/3 0 1 2, 1/3 0 0 2 2
MODULATION = 3 3 1, 1/3 1/3 0 1 1 0, 1/3 1/3 0 1 1 90
MODULATION = -1 1 1 1 -1 1 1 1 -1, 1/2 1/2 0 1 2
Characters of irreducible representations#
IRREPS#
Characters of irreducible representations (Irreps) of phonon modes are shown.
For this calculation, a primitive cell has to be used. If the input unit cell is
a non-primitive cell, it has to be transformed to a primitive cell using
PRIMITIVE_AXES tag.
The first three values gives a q-point in reduced coordinates to be calculated. The degenerated modes are searched only by the closeness of frequencies. The frequency difference to be tolerated is specified by the fourth value in the frequency unit that the user specified.
IRREPS = 0 0 0 1e-3
Symbols of Irreps for the 32 point group types at the \(\Gamma\) point are shown but not at non-\(\Gamma\) point.
SHOW_IRREPS#
Irreducible representations are shown along with character table.
IRREPS = 1/3 1/3 0
SHOW_IRREPS = .TRUE.
LITTLE_COGROUP#
Show irreps of little co-group (point-group of wavevector) instead of little group.
IRREPS = 0 0 1/8
LITTLE_COGROUP = .TRUE.
Input/Output file control#
FC_FORMAT, READFC_FORMAT, WRITEFC_FORMAT#
There are two file-formats to store force constants. Currently
text style (TEXT) and hdf5 (HDF5) formats are
supported. The default file format is the
text style. Reading and writing force constants are
invoked by FORCE_CONSTANTS tag. Using these tags,
the input/output formats are switched.
FC_FORMAT affects to both input and output, e.g.
FORCE_CONSTANTS = WRITE
FC_FORMAT = HDF5
READFC_FORMAT and WRITEFC_FORMAT can be used to control input and output
formats separately, i.e., the following setting to convert force constants
format is possible:
READ_FORCE_CONSTANTS = .TRUE.
WRITE_FORCE_CONSTANTS = .TRUE.
WRITEFC_FORMAT = HDF5
BAND_FORMAT, MESH_FORMAT, QPOINTS_FORMAT#
There are two file-formats to write the results of band structure, mesh, and
q-points calculations. Currently YAML (YAML) and hdf5 (HDF5) formats are
supported. The default file format is the YAML format. The file format is
changed as follows:
BAND_FORMAT#
When HDF5 is set, calculated result is stored in band.hdf5 instead of
band.yaml.
BAND_FORMAT = HDF5
MESH_FORMAT#
When HDF5 is set, calculated result is stored in mesh.hdf5 instead of
mesh.yaml.
MESH_FORMAT = HDF5
QPOINTS_FORMAT#
When HDF5 is set, calculated result is stored in qpoints.hdf5 instead of
qpoints.yaml.
QPOINTS_FORMAT = HDF5
HDF5#
The following output files are written in hdf5 format instead of their original
formats (in parenthesis) by HDF5 = .TRUE.. In addition, force_constants.hdf5
is read with this tag.
force_constants.hdf5(FORCE_CONSTANTS)mesh.hdf5(mesh.yaml)band.hdf5(band.yaml)qpoints.hdf5(qpoints.yaml)
HDF5 = .TRUE.
force_constants.hdf5#
With --hdf5 option and FORCE_CONSTANTS = WRITE (--writefc),
force_constants.hdf5 is written. With --hdf5 option and
FORCE_CONSTANTS = READ (--readfc), force_constants.hdf5 is read.
mesh.hdf5#
In the mesh sampling calculations (see Mesh sampling tags), calculation
results are written into mesh.hdf5 but not into mesh.yaml. Using this option
may reduce the data output size and thus writing time when mesh.yaml is huge,
e.g., eigenvectors are written on a dense sampling mesh.
qpoints.hdf5#
In the specific q-points calculations (QPOINTS), calculation results
are written into qpoints.hdf5 but not into qpoints.yaml. With
WRITEDM, dynamical matrices are also stored in qpoints.hdf5. Using
this option may be useful with large set of q-points with including eigenvector
or dynamical matrix output.
band.hdf5#
In the band structure calculations (Band structure tags),
calculation results are written into band.hdf5 but not into band.yaml.
#
The following data may be optionally included in the phonopy.yaml file that is
written at the end of the pre/post-process (after running the phonopy script).
INCLUDE_FC#
The --include-fc flag or setting INCLUDE_FC = .TRUE. will cause the force
constants (if available) to be written as an entry in phonopy.yaml file.
The written force constants will reflect the required/available format used
during processing. So if --full-fc is set the entire matrix will be written.
INCLUDE_FS#
The --include-fs flag or setting INCLUDE_FS = .TRUE. will cause the force
sets (if available) to be written as an entry in phonopy.yaml.
INCLUDE_DISP#
The --include-disp flag or setting INCLUDE_DISP = .TRUE. will cause
displacements data (if available) to be written as an entry in phonopy.yaml
file.
INCLUDE_ALL#
All available data covered by the other include flags can be written to
phonopy.yaml file using the --include-all flag or by setting INCLUDE_ALL = .TRUE.. Force constants are not stored when force sets are stored.