Command options / Setting tags#
Use of configuration file#
Phono3py is operated with command options or with a configuration file that contains setting tags. In this page, the command options are explained. Most of command options have their respective setting tags.
A configuration file with setting tags like phonopy can be used instead of and
together with the command options. The setting tags are mostly equivalent to the
respective most command options, but when both are set simultaneously, the
command options are preferred. An example of configuration (e.g., saved in a
file setting.conf) is as follow:
DIM = 2 2 2
DIM_FC2 = 4 4 4
PRIMITIVE_AXES = 0 1/2 1/2 1/2 0 1/2 1/2 1/2 0
MESH = 11 11 11
BTERTA = .TRUE.
NAC = .TRUE.
READ_FC2 = .TRUE.
READ_FC3 = .TRUE.
CELL_FILENAME = POSCAR-unitcell
where the setting tag names are case insensitive. This is run by
% phono3py setting.conf [OPTIONS]
or
% phono3py [OPTIONS] -- setting.conf
When using phono3py (see also phono3py command)
% phono3py --config setting.conf [OPTIONS]
Input cell file name#
-c (CELL_FILENAME)#
This specifies input unit cell filename.
% phono3py-init -c POSCAR-unitcell [OPTIONS]
Calculator interface#
--qe (CALCULATOR = QE)#
Quantum espresso (pw) interface is invoked. See the detail at Quantum ESPRESSO (pw) & phono3py calculation.
--crystal (CALCULATOR = CRYSTAL)#
CRYSTAL interface is invoked. See the detail at CRYSTAL & phono3py calculation.
--turbomole (CALCULATOR = TURBOMOLE)#
TURBOMOLE interface is invoked. See the details at TURBOMOLE & phono3py calculation.
Utilities to create default input files#
These options have no respective configuration file tags.
--cf3 (command option only)#
This is used to create FORCES_FC3 from phono3py_disp.yaml and force
calculator outputs containing forces in supercells. phono3py_disp.yaml has to
be located at the current directory.
% phono3py-init --cf3 disp-{00001..00755}/vasprun.xml
% phono3py-init --cf3 supercell_out/disp-{00001..00111}/Si.out
Note
The calculator interface should be stored in phono3py_disp.yaml, so it is not
needed to set it manually. Command-line-options like --qe will be ignored. If
the calculator interface is missing from phono3py_disp.yaml but needed, please
update the phono3py section in the file as follows:
phono3py:
calculator: qe
--cf3-file (command option only)#
This is used to create FORCES_FC3 from a text file containing a list of
calculator output file names. phono3py_disp.yaml has to be located at the
current directory. The calculator interface is unnecessary to specify, see the
note at –cf3.
% phono3py-init --cf3-file file_list.dat
where file_list.dat contains file names that can be recognized from the
current directory and is expected to be like:
disp-00001/vasprun.xml
disp-00002/vasprun.xml
disp-00003/vasprun.xml
disp-00004/vasprun.xml
...
The order of the file names is important. This option may be useful to be used
together with --cutoff-pair option.
--cf2 (command option only)#
This is used to create FORCES_FC2 similarly to --cf3 option.
phono3py_disp.yaml has to be located at the current directory. This is
optional. FORCES_FC2 is necessary to run with --dim-fc2 option. The
calculator interface is unnecessary to specify, see the note at –cf3.
% phono3py-init --cf2 disp_fc2-{00001..00002}/vasprun.xml
--cfz (command option only)#
This is used to create FORCES_FC3 and FORCES_FC2 subtracting residual forces
combined with --cf3 and --cf2, respectively. The calculator interface is
unnecessary to specify, see the note at –cf3.
In the following example, it is supposed that disp3-00000/vasprun.xml and
disp2-00000/vasprun.xml contain the forces of the perfect supercells. In ideal
case, these forces are zero, but often they are not. Here, this is called
“residual forces”. Sometimes quality of force constants is improved in this way.
% phono3py-init --cf3 disp3-{00001..01254}/vasprun.xml --cfz disp3-00000/vasprun.xml
% phono3py-init --cf2 disp2-{00001..00006}/vasprun.xml --cfz disp2-00000/vasprun.xml
--fs2f2 or --force-sets-to-forces-fc2 (command option only)#
FORCES_FC2 is created from phonopy’s FORCE_SETS file. Necessary yaml lines
for phono3py_disp.yaml is displayed as text.
% phono3py-init --fs2f2
--cfs or --create-force-sets (command option only)#
Phonopy’s FORCE_SETS is created from FORCES_FC3 and phono3py_disp.yaml.
% phono3py-init --cfs
In conjunction with –dim-fc2, phonopy’s FORCE_SETS is
created from FORCES_FC2 and phono3py_disp.yaml instead of FORCES_FC3 and
phono3py_disp.yaml.
% phono3py-init --cfs --dim-fc2 4 4 4
--sp or --save-params#
Instead of FORCES_FC3, phono3py_params.yaml is generated. This option must
be used with --cf3, and optionally with --cf2. If the force calculator
supports reading energy of supercell, those are written into
phono3y_params.yaml. These energies are necessary for using --pypolymlp
option.
% phono3py-init --cf3 disp-{00001..00755}/vasprun.xml --sp
When using with --cf2, --cf3 has to be specified simultaneously as below,
% phono3py-init --cf3 disp-{00001..00755}/vasprun.xml --cf2 disp_fc2-{00001..00002}/vasprun.xml --sp
Supercell, primitive cell, masses, magnetic moments#
--dim (DIM)#
phono3py doesn’t have this option.
Supercell dimension is specified. See the detail at
http://phonopy.github.io/phonopy/setting-tags.html#dim. When
phono3py_disp.yaml is found in the current directory, it is read
automatically. Since supercell dimension is written in this file, --dim is
unnecessary to specify. For example, just
% phono3py --fc-symmetry
or
% phono3py --symfc
can be used to calculate force constants.
--dim-fc2 (DIM_FC2)#
phono3py doesn’t have this option.
Supercell dimension for 2nd order force constants (for harmonic phonons) is
specified. This is optional. When a proper phono3py_disp.yaml exists in the
current directory, this is unnecessary to be specified.
A larger and different supercell size for 2nd order force constants than that for 3rd order force constants can be specified with this option. Often interaction between a pair of atoms has longer range in real space than interaction among three atoms. Therefore to reduce computational demand, choosing larger supercell size only for 2nd order force constants may be a good idea.
Using this option with -d option, the structure files (e.g. POSCAR_FC2-xxxxx
or equivalent files for the other interfaces) and phono3py_disp.yaml are
created. These are used to calculate 2nd order force constants for the larger
supercell size and these force calculations have to be done in addition to the
usual force calculations for 3rd order force constants.
% phono3py-init -d --dim 2 2 2 --dim-fc2 4 4 4 --pa auto -c POSCAR-unitcell
After the force calculations, --cf2 option is used to create FORCES_FC2.
% phono3py-init --cf2 disp-{001,002}/vasprun.xml
To calculate 2nd order force constants for the larger supercell size,
FORCES_FC2 and phono3py_disp.yaml are necessary. Whenever running phono3py
for the larger 2nd order force constants, --dim-fc2 option has to be
specified. fc2.hdf5 created as a result of running phono3py contains the 2nd
order force constants with larger supercell size. The filename is the same as
that created in the usual phono3py run without --dim-fc2 option.
% phono3py --dim 2 2 2 --dim_fc2 4 4 4 -c POSCAR-unitcell [OPTIONS]
--pa, --primitive-axes (PRIMITIVE_AXES)#
Transformation matrix from a non-primitive cell to the primitive cell. See
phonopy PRIMITIVE_AXES tag (--pa option) at primitive-axis
(phonopy).
When phono3py_disp.yaml contains this information and phono3py_disp.yaml is
read when running phono3py or phono3py command, this is unnecessary to
be specified.
--mass (MASS)#
Atomic masses of primitive cell are overwritten. See more details in phonopy web page.
--magmom (MAGMOM)#
Magnetic moments of unit cell are specified. This information is used to find crystal symmetry. See more details in phonopy web page.
Displacement creation#
-d (CREATE_DISPLACEMENTS = .TRUE.)#
phono3py doesn’t have this option.
Supercells with displacements and phono3py_disp.yaml are created. Using with
--amplitude option, atomic displacement distances are controlled. With this
option, files for supercells with displacements and phono3py_disp.yaml file
are created.
It is recommended to use this option with --pa auto option to store
information about primitive cell (primitive_matrix key) in
phono3py_disp.yaml, e.g.,
% phono3py-init -c POSCAR-unitcell -d --dim 2 2 2 --dim-fc2 4 4 4 --pa auto
--rd (RANDOM_DISPLACEMENTS), --rd-fc2 (RANDOM_DISPLACEMENTS_FC2) and --random-seed (RANDOM_SEED)#
See also Force constants calculation with random displacements of atoms.
Random directional displacements are generated for fc3 and fc2 supercells by
--rd and --rd-fc2, respectively. --rd auto can estimate a possible number
of supercells required (see --rd-auto-factor (RD_NUMBER_ESTIMATION_FACTOR)).
--amplitude and --random-seed options may be used together. These are used
in the equivalent way to --rd of
phonopy.
Like -d option, it is recommended to specify --pa auto together with --rd
and/or --rd-fc2,
% phono3py-init -c POSCAR-unitcell --dim 2 2 2 --dim-fc2 4 4 4 --rd 100 --rd-fc2 2 --pa auto
--rd-auto-factor (RD_NUMBER_ESTIMATION_FACTOR)#
This scales the number of supercells generated by --rd auto by the specified
factor.
--amplitude (DISPLACEMENT_DISTANCE)#
phono3py doesn’t have this option.
Atomic displacement distance is specified. This value may be increased for the weak interaction systems and decreased when the force calculator is numerically very accurate.
The default value depends on calculator. See Default displacement distance created.
--fc-calc, --fc-calculator (FC_CALCULATOR)#
Choice of force constants calculator.
% phono3py --fc-calc symfc ...
To use different force constants calculators for fc2 and fc3
% phono3py --fc-calc "symfc|" ...
Those for fc2 and fc3 are separated by | such as symfc| . Blank means to
employ the finite difference method for systematic displacements generated by
the option -d.
--fc-calc-opt, --fc-calculator-options (FC_CALCULATOR_OPTIONS)#
Special options for force constants calculators.
% phono3py --fc-calc-opt "cutoff=8" ...
Similarly to --fc-calc, | can be used to separated those for fc2 and fc3.
Options for symfc#
cutoff : cutoff pair distance beyond that third-order force constants are zero (fc3 only).
use_mkl : sparse_dot_mkl is employed when it is available.
--symfc and --alm#
These are shortcuts of --fc-calc symfc and --fc-calc alm, respectively.
Please be careful that --symfc and --sym-fc (deprecated) are similar, but
different.
Refer to the symfc and alm sections in the Phonopy documentation for additional details.
Force constants#
--full-fc (COMPACT_FC = .FALSE.)#
When creating force constants from FORCES_FC3 and/or FORCES_FC2, the
compact-array format is used by default. The shape of the data array is
(num_patom, num_satom) for fc2 and (num_patom, num_satom, num_satom) for
fc3, where num_patom and num_satom are the numbers of atoms in the primitive
cell and supercell. With --full-fc, the full-size arrays are computed instead,
where num_patom is replaced by num_satom. The compact form reduces data
size, and the reduction grows with supercell dimension. If the input crystal
structure has centring, –pa is necessary to have the
smallest data size. In this case, --pa option has to be specified on reading.
Otherwise phono3py can recognize if fc2.hdf5 and fc3.hdf5 are compact or
full automatically. When using with --fc-symmetry, the calculated results will
become slightly different due to the imperfect symmetrization scheme that
phono3py employs.
% phono3py --full-fc
--cfc or --compact-fc (COMPACT_FC = .TRUE.) (deprecated)#
Compact force constants are now the default, so this option has no effect and
is retained only for backward compatibility. Specifying --cfc or
--compact-fc on the command line emits a deprecation notice. Use --full-fc
to opt back into the full-array format.
--fc-symmetry (FC_SYMMETRY = .TRUE.)#
Second- and third-order force constants are symmetrized. The index exchange of real space force constants and translational invariance symmetry are applied in a simple way. This symmetrization just removes drift force constants evenly from all elements and then applies averaging index-exchange equivalent elements. Therefore the different symmetries are not simultaneously enforced. For better symmetrization, it is recommended to use an external force constants calculator like ALM.
The symmetrizations for the second and third orders can be independently applied
by --sym-fc2 (SYMMETRIZE_FC2 = .TRUE.) and --sym-fc3r
(SYMMETRIZE_FC3 = .TRUE.), , respectively.
--symfc-projector (USE_SYMFC_PROJECTOR = .TRUE.)#
Symmetrize force constants using the symfc projector instead of the traditional
approach. This applies both when force constants are computed from displacements
and when they are read from existing fc3.hdf5 / fc2.hdf5 files. In the
latter case, the symmetrized force constants are written back to those files.
% phono3py phono3py.yaml --symfc-projector
--cutoff-fc3 or --cutoff-fc3-distance (CUTOFF_FC3_DISTANCE)#
This option is not used to reduce number of supercells with displacements, but this option is used to set zero in elements of given third-order force constants. The zero elements are selected by the condition that any pair-distance of atoms in each atom triplet is larger than the specified cut-off distance.
If one wants to reduce number of supercells, the first choice is to reduce the
supercell size and the second choice is using --cutoff-pair option.
--cutoff-pair or --cutoff-pair-distance (CUTOFF_PAIR_DISTANCE)#
This option works in two ways.
When using with -d options, a cutoff pair-distance in a supercell is used to
reduce the number of necessary supercells with displacements to obtain third
order force constants. As the drawback, a certain number of
third-order-force-constants elements are abandoned or computed with less
numerical accuracy. More details are found at Force constants calculation with cutoff pair-distance.
When using with an external force constants calculator, --cutoff-pair VAL works
equivalent to --fc-calc-opt "cutoff=VAL".
--alm#
This invokes ALM as the force constants calculator for fc2 and fc3. See the
detail at
phonopy documentation.
This option is useful for fitting random displacement dataset or MD data to
force constants. Phono3py doesn’t provide command-line interface to generate
random displacements. Instead simply
phonopy can be used for this purpose,
because FORCE_SETS in the type-II format obtained using phonopy can be used as
FORCES_FC3 and FORCES_FC2 just renaming the file name.
Reciprocal space sampling mesh and grid points, and band indices#
--mesh (MESH or MESH_NUMBERS)#
Mesh sampling grids in reciprocal space are generated with the specified numbers. This mesh is made along reciprocal axes and is always Gamma-centered. Except for that this mesh is always Gamma-centered, this works in the same way as written here.
--gp (GRID_POINTS)#
Grid points are specified by their unique indices, e.g., for selecting the q-points where imaginary parts of self energies are calculated. For thermal conductivity calculation, this can be used to distribute its calculation over q-points (see Workload distribution).
Indices of grid points are specified by space or comma (,) separated numbers.
The mapping table between grid points to its indices is obtained by running with
--loglevel=2 option.
% phono3py --mesh 19 19 19 --br --write-gamma --gp 0 1 2 3 4 5
where --gp 0 1 2 3 4 5 can be also written --gp="0,1,2,3,4,5". --ga
option below can be used similarly for the same purpose.
--ga (GRID_ADDRESSES)#
This is used to specify grid points like --gp option but in their addresses
represented by integer numbers. For example with --mesh 16 16 16, a q-point
of (0.5, 0.5, 0.5) is given by --ga 8 8 8. The values have to be integers.
If you want to specify the point on a path,
--ga 0 0 0 1 1 1 2 2 2 3 3 3 ..., where each three values are recognized as
a grid point. The grid points given by --ga option are translated to grid
point indices as given by --gp option, and the values given by --ga option
will not be shown in log files.
--bi (BAND_INDICES)#
Band indices are specified. The calculated values at indices separated by space
are averaged, and those separated by comma are separately calculated. The output
file name will be, e.g., gammas-mxxx-gxx(-sx)-bx.dat where bxbx... shows the
band indices where the values are calculated and summed and averaged over those
bands.
% phono3py --mesh 16 16 16 --gp 34 --bi "4 5, 6"
This option may be also useful to distribute the computational demand such like that the unit cell is large and the calculation of phonon-phonon interaction is heavy.
--wgp (command option only)#
Irreducible grid point indices and related information are written into
ir_grid_points.yaml. This information may be used when we want to distribute
thermal conductivity calculation into small pieces or to find specific grid
points to calculate imaginary part of self energy, for which
–gp option can be used to specify the grid point indices.
grid_address-mxxx.hdf5 is also written. This file contains all the grid points
and their grid addresses in integers. Q-points corresponding to grid points are
calculated divided these integers by sampling mesh numbers for respective
reciprocal axes.
% phono3py --mesh 19 19 19 --wgp
--stp (command option only)#
Numbers of q-point triplets to be calculated for irreducible grid points for
specified sampling mesh numbers are shown. This can be used to estimate how
large a calculation is. Only those for specific grid points are shown by using
with --gp or --ga option.
% phono3py --mesh 19 19 19 --stp --gp 20
Brillouin zone integration#
--thm (TETRAHEDRON = .TRUE.)#
Tetrahedron method is used for calculation of imaginary part of self energy. This is the default option. Therefore it is not necessary to specify this unless both results by tetrahedron method and smearing method in one time execution are expected.
--sigma (SIGMA)#
\(\sigma\) value of Gaussian function for smearing when calculating imaginary part of self energy.
Multiple \(\sigma\) values are also specified by space separated numerical values. This is used when we want to test several \(\sigma\) values simultaneously.
--sigma-cutoff (SIGMA_CUTOFF_WIDTH)#
The tails of the Gaussian functions that are used to replace delta functions in
the equation shown at –full-pp are cut with this
option. The value is specified in number of standard deviation.
--sigma-cutoff 5 gives the Gaussian functions to be cut at \(5\sigma\). Using
this option scarifies the numerical accuracy. So the number has to be carefully
tested. But computation of phonon-phonon interaction strength becomes much
faster in exchange for it.
--full-pp (FULL_PP = .TRUE.)#
For thermal conductivity calculation using the linear tetrahedron method (from
version 1.10.5) and smearing method with --simga-cutoff (from version 1.12.3),
only necessary elements (i.e., that have non-zero delta functions) of
phonon-phonon interaction strength,
\(\bigl|\Phi_{-\lambda\lambda'\lambda''}\bigl|^2\), is calculated due to delta
functions in calculation of \(\Gamma_\lambda(\omega)\),
But using this option, full elements of phonon-phonon interaction strength are calculated and averaged phonon-phonon interaction strength (\(P_{\mathbf{q}j}\), see –ave-pp) is also given and stored.
Methods to solve phonon Boltzmann equation and inter-band transport#
--br (BTERTA = .TRUE.)#
Run calculation of lattice thermal conductivity tensor with the single mode
relaxation time approximation (RTA) and linearized phonon Boltzmann equation.
Without specifying --gp (or --ga) option, all necessary phonon lifetime
calculations for grid points are sequentially executed and then thermal
conductivity is calculated under RTA. The thermal conductivity and many related
properties are written into kappa-mxxx.hdf5.
With --gp (or --ga) option, phonon lifetimes on the specified grid points
are calculated. To save the results, --write-gamma option has to be specified
and the physical properties belonging to the grid points are written into
kappa-mxxx-gx(-sx).hdf5.
--lbte (LBTE = .TRUE.)#
Run calculation of lattice thermal conductivity tensor with a direct solution of
linearized phonon Boltzmann equation. The basis usage of this option is
equivalent to that of --br. More detail is documented at
Direct solution of linearized phonon Boltzmann equation.
--tt, --transport-type (TRANSPORT_TYPE)#
Select the transport formulation used to evaluate the inter-band
contributions to the thermal conductivity. This option is combined with --br
(RTA) or --lbte (direct solution). Without --tt, the standard particle-like
formulation is used. The built-in variants njc23, ibdb19, and smm19 are
experimental.
wte invokes the external phono3py-wte plugin. More detail is documented at
Inter-band transport.
Scattering#
--isotope (ISOTOPE =.TRUE.)#
Phonon-isotope scattering is calculated based on the formula by Shin-ichiro Tamura, Phys. Rev. B, 27, 858 (1983). Mass variance parameters are read from database of the natural abundance data for elements, which refers Laeter et al., Pure Appl. Chem., 75, 683 (2003).
% phono3py -v --mesh 32 32 20 --br --isotope
--mass-variances or --mv (MASS_VARIANCES)#
Mass variance parameters are specified by this option to include phonon-isotope
scattering effect in the same way as --isotope option. For example of GaN,
this may be set like --mv 1.97e-4 1.97e-4 0 0. The number of elements has to
correspond to the number of atoms in the primitive cell.
Isotope effect to thermal conductivity may be checked first running without isotope calculation:
% phono3py -v --mesh 32 32 20 --br
Then running with isotope calculation:
% phono3py -v --mesh 32 32 20 --br --read-gamma --mv 1.97e-4 1.97e-4 0 0
In the result hdf5 file, currently isotope scattering strength is not written
out, i.e., gamma is still imaginary part of self energy of ph-ph scattering.
--boundary-mfp, --bmfp (BOUNDARY_MFP)#
A most simple phonon boundary scattering treatment is included. \(v_g/L\) is just used as the scattering rate, where \(v_g\) is the group velocity and \(L\) is the boundary mean free path. The value is given in micrometer. The default value, 1 metre, is just used to avoid divergence of phonon lifetime and the contribution to the thermal conductivity is considered negligible.
--ave-pp (USE_AVE_PP = .TRUE.)#
Averaged phonon-phonon interaction strength (\(P_{\mathbf{q}j}=P_\lambda\)) is used to calculate imaginary part of self energy in thermal conductivity calculation. \(P_\lambda\) is defined as
where \(n_\text{a}\) is the number of atoms in unit cell. This is roughly constant with respect to the sampling mesh density for converged \(|\Phi_{\lambda \lambda' \lambda''}|^2\). Then for all \(\mathbf{q}',j',j''\),
where \(N\) is the number of grid points on the sampling mesh. \(\Phi_{\lambda \lambda' \lambda''} \equiv 0\) unless \(\mathbf{q} + \mathbf{q}' + \mathbf{q}'' = \mathbf{G}\).
See also reference papers.
This option works only when --read-gamma and --br options are activated
where the averaged phonon-phonon interaction that is read from
kappa-mxxx(-sx-sdx).hdf5 file is used if it exists in the file. Therefore the
averaged phonon-phonon interaction has to be stored before using this option
(see –full-pp). The calculation result overwrites
kappa-mxxx(-sx-sdx).hdf5 file. Therefore the original
kappa-mxxx(-sx-sdx).hdf5 file should be backed up.
First, run full conductivity calculation,
% phono3py -v --mesh 32 32 20 --br
Then
% phono3py -v --mesh 32 32 20 --br --read-gamma --ave-pp -o ave_pp
--const-ave-pp (CONST_AVE_PP = .TRUE.)#
Averaged phonon-phonon interaction (\(P_{\mathbf{q}j}\)) is replaced by this
constant value and \(|\Phi_{\lambda \lambda'
\lambda''}|^2\) are set as written in
–ave-pp for thermal conductivity calculation. This
option works only when --br options are activated. Therefore third-order force
constants are not necessary to input. The physical unit of the value is
\(\text{eV}^2\).
See also reference papers.
% phono3py -v --mesh 32 32 20 --br --const-ave-pp 1e-10
--nu (N_U = .TRUE.)#
Integration over q-point triplets for the calculation of \(\Gamma_\lambda(\omega_\lambda)\) is made separately for normal \(\Gamma^\text{N}_\lambda(\omega_\lambda)\) and Umklapp \(\Gamma^\text{U}_\lambda(\omega_\lambda)\) processes. The sum of them is usual \(\Gamma_\lambda(\omega_\lambda) = \Gamma^\text{N}_\lambda(\omega_\lambda) + \Gamma^\text{U}_\lambda(\omega_\lambda)\) and this is used to calculate thermal conductivity in single-mode RTA. The separation, i.e., the choice of G-vector, is made based on the first Brillouin zone. See gamma_N and gamma_U.
--scattering-event-class (SCATTERING_EVENT_CLASS)#
Scattering event class of imaginary part of self energy is specified by 1 or
2. This only works with --ise (IMAG_SELF_ENERGY = .TRUE.) option. The classes 1 and 2 are
given by
and
respectively, and
Temperature#
--ts (TEMPERATURES): Temperatures#
Specific temperatures are specified by --ts.
% phono3py -v --mesh 11 11 11 -c POSCAR-unitcell --br --ts 200 300 400
--tmax, --tmin, --tstep (TMAX, TMIN, TSTEP)#
Temperatures at equal interval are specified by --tmax, --tmin, --tstep.
See phonopy’s document for the same tags at
http://phonopy.github.io/phonopy/setting-tags.html#tprop-tmin-tmax-tstep.
% phono3py -v --mesh 11 11 11 --br --tmin 100 --tmax 1000 --tstep 50
Non-analytical term correction#
--nac (NAC = .TRUE.)#
Non-analytical term correction for harmonic phonons. Like as phonopy, BORN
file has to be put on the same directory. Always the default value of unit
conversion factor is used even if it is written in the first line of BORN
file.
--q-direction (Q_DIRECTION)#
This is used with --nac to specify reciprocal-space direction at
\(\mathbf{q}\rightarrow \mathbf{0}\). See the detail at
http://phonopy.github.io/phonopy/setting-tags.html#q-direction.
Imaginary and real parts of self energy#
Phonon self-energy of bubble diagram is written as,
The imaginary part and real part are written as
and
respectively. In the above formulae, angular frequency \(\omega\) is used, but in the calculation results, ordinal frequency \(\nu\) is used. Be careful about \(2\pi\) treatment.
See also reference papers.
--ise (IMAG_SELF_ENERGY = .TRUE.)#
Imaginary part of self energy \(\Gamma_\lambda(\omega)\) is calculated with
respect to frequency \(\omega\), where \(\omega\) is sampled following
--num-freq-points, --freq-pitch (NUM_FREQUENCY_POINTS). The output of \(\Gamma_\lambda(\omega)\) is written
to gammas-mxxx-gx(-sx)-tx-bx.dat in THz (without \(2\pi\)) with respect to
samplied frequency points of \(\omega\) in THz (without \(2\pi\)).
% phono3py --mesh 16 16 16 --q-direction 1 0 0 --gp 0 --ise --bi "4 5, 6"
--rse (REAL_SELF_ENERGY = .TRUE.)#
Real part of self energy \(\Delta_\lambda(\omega)\) is calculated with respect to
frequency \(\omega\), where \(\omega\) is sampled following
--num-freq-points, --freq-pitch (NUM_FREQUENCY_POINTS). With this option, only smearing approach is
provided, for which values given by --sigma option are used to approximate the
principal value as \(\varepsilon\) in the following equation:
where \(\mathcal{P}\) denotes the Cauchy principal value. The output of
\(\Delta_\lambda(\omega)\) is written to deltas-mxxx-gx-sx-tx-bx.dat in THz
(without \(2\pi\)) with respect to samplied frequency points of \(\omega\) in THz
(without \(2\pi\)).
% phono3py --mesh 16 16 16 --q-direction 1 0 0 --gp 0 --rse --sigma 0.1 --bi "4 5, 6"
Spectral function#
Phonon spectral function of bubble diagram is written as
where \(A_\lambda(\omega)\) is defined to be normalized as
See also reference papers.
--spf (SPECTRAL_FUNCTION = .TRUE.)#
Spectral function of self energy \(A_\lambda(\omega)\) is calculated with respect
to frequency \(\omega\), where \(\omega\) is sampled following
--num-freq-points, --freq-pitch (NUM_FREQUENCY_POINTS). First, imaginary part of self-energy is calculated
and then the real part is calculated using the Kramers–Kronig relation. The
output of \(A_\lambda(\omega)\) is written to spectral-mxxx-gx(-sx)-tx-bx.dat in
THz (without \(2\pi\)) with respect to samplied frequency points of \(\omega\) in
THz (without \(2\pi\)), and spectral-mxxx-gx.hdf5.
% phono3py --mesh 16 16 16 --q-direction 1 0 0 --gp 0 --spf
Note
When --bi option is unspecified, spectral functions of all bands are
calculated and the sum divided by the number of bands is stored in
spectral-mxxx-gx(-sx)-tx-bx.dat, i.e.,
\((\sum_j A_{\mathbf{q}j}) / N_\text{b}\), where \(N_\text{b}\) is the
number of bands and \(\lambda \equiv (\mathbf{q},j)\) is the phonon mode.
The spectral function of each band is written in the hdf5
file, where \(A_{\mathrm{q}j}\) is normalied as given above, i.e., numerical
sum of stored value for each band should become roughly 1.
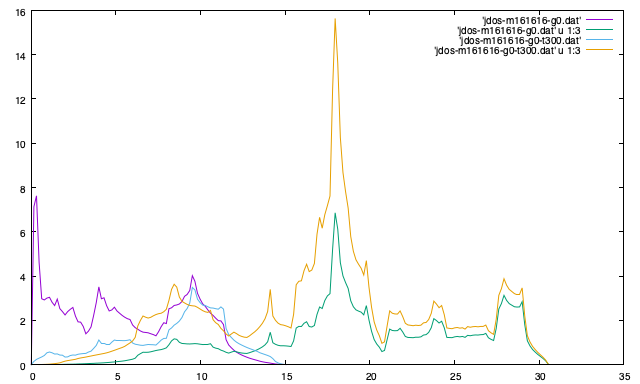
Joint density of states (JDOS) and weighted-JDOS#
--jdos (JOINT_DOS = .TRUE.)#
Two classes of joint density of states (JDOS) are calculated. The result is
written into jdos-mxxx-gx(-sx-sdx).dat in \(\text{THz}^{-1}\) (without
\((2\pi)^{-1}\)) with respect to frequency in THz (without \(2\pi\)). Frequency
sampling points can be specified by --num-freq-points, --freq-pitch (NUM_FREQUENCY_POINTS).
The first column is the frequency, and the second and third columns are the values given as follows, respectively,
See also reference papers.
% phono3py --mesh 16 16 16 --jdos --ga 0 0 0 8 8 8
When temperatures are specified, two classes of weighted JDOS are calculated.
The result is written into jdos-mxxx-gx(-sx)-txxx.dat in \(\text{THz}^{-1}\)
(without \((2\pi)^{-1}\)) with respect to frequency in THz (without \(2\pi\)). In
the file name, txxx shows the temperature. The first column is the frequency,
and the second and third columns are the values given as follows, respectively,
See also reference papers.
% phono3py --mesh 16 16 16 --jdos --ga 0 0 0 8 8 8 --ts 300
This is an example of Si-PBEsol.
Sampling frequency for distribution functions#
--num-freq-points, --freq-pitch (NUM_FREQUENCY_POINTS)#
For spectrum-like calculations of imaginary part of self energy, spectral
function, and JDOS, number or interval of uniform sampling frequency points is
controlled by --num-freq-points or --freq-pitch. Both are unspecified,
default value of --num-freq-points of 200 is used.
Mode-Gruneisen parameter from 3rd order force constants#
--gruneisen (GRUNEISEN = .TRUE.)#
Mode-Gruneisen-parameters are calculated from fc3.
Mesh sampling mode:
% phono3py -v --mesh 16 16 16 --gruneisen
Band path mode:
% phono3py -v --gruneisen --band "0 0 0 0 0 1/2"
File I/O#
--fc2 (READ_FC2 = .TRUE.)#
Read 2nd order force constants from fc2.hdf5.
--fc3 (READ_FC3 = .TRUE.)#
Read 3rd order force constants from fc3.hdf5.
--write-gamma (WRITE_GAMMA = .TRUE.)#
Imaginary parts of self energy at harmonic phonon frequencies
\(\Gamma_\lambda(\omega_\lambda)\) are written into file in hdf5 format. The
result is written into kappa-mxxx-gx(-sx-sdx).hdf5 or
kappa-mxxx-gx-bx(-sx-sdx).hdf5 with --bi option. With --sigma and
--sigma-cutoff options, -sx and --sdx are inserted, respectively, in front
of .hdf5.
--read-gamma (READ_GAMMA = .TRUE.)#
Imaginary parts of self energy at harmonic phonon frequencies
\(\Gamma_\lambda(\omega_\lambda)\) are read from kappa file in hdf5 format.
Initially the usual result file of kappa-mxxx(-sx-sdx).hdf5 is searched.
Unless it is found, it tries to read kappa file for each grid point,
kappa-mxxx-gx(-sx-sdx).hdf5. Then, similarly, kappa-mxxx-gx(-sx-sdx).hdf5
not found, kappa-mxxx-gx-bx(-sx-sdx).hdf5 files for band indices are searched.
--write-gamma-detail (WRITE_GAMMA_DETAIL = .TRUE.)#
Each q-point triplet contribution to imaginary part of self energy is written
into gamma_detail-mxxx-gx(-sx-sdx).hdf5 file. Be careful that this can be a
large file. See gamma_detail-*.hdf5.
--write-phonon (WRITE_PHONON = .TRUE.)#
Phonon frequencies, eigenvectors, and grid point addresses are stored in
phonon-mxxx.hdf5 file. –pa and –nac
may be required depending on calculation setting. See phonon-*.hdf5.
% phono3py --mesh 11 11 11 --write-phoonon
--read-phonon (READ_PHONON = .TRUE.)#
Phonon frequencies, eigenvectors, and grid point addresses are read from
phonon-mxxx.hdf5 file and the calculation is continued using these phonon
values. This is useful when we want to use fixed phonon eigenvectors that can be
different for degenerate bands when using different eigenvalue solvers or
different CPU architectures. –pa and
–nac may be required depending on calculation setting.
% phono3py --mesh 11 11 11 --read-phoonon --br
--write-pp (WRITE_PP = .TRUE.) and --read-pp (READ_PP = .TRUE.)#
Phonon-phonon (ph-ph) interaction strengths are written to and read from
pp-mxxx-gx.hdf5. This works only in the calculation of lattice thermal
conductivity, i.e., usable only with --br or --lbte. The stored data are
different with and without specifying --full-pp option. In the former case,
all the ph-ph interaction strengths among considered phonon triplets are stored
in a simple manner, but in the later case, only necessary elements to calculate
collisions are stored in a complicated way. In the case of RTA conductivity
calculation, in writing and reading, ph-ph interaction strength has to be stored
in memory, so there is overhead in memory than usual RTA calculation.
% phono3py --mesh 11 11 11 --write-pp --br --gp 1
% phono3py --mesh 11 11 11 --read-pp --br --gp 1
--hdf5-compression (command option only)#
Most of phono3py HDF5 output file is compressed by default with the gzip
compression filter. To avoid compression, --hdf5-compression=None has to be
set. Other filters (lzf or integer values of 0 to 9) may be used, see h5py
documentation
(http://docs.h5py.org/en/stable/high/dataset.html#filter-pipeline).