CRYSTAL & phonopy calculation#
CRYSTAL program package has a robust built-in phonon calculation workflow. However, the Phonopy interface enables convenient access to many phonon-related properties, such as subsequent Phono3py calculations of lattice thermal conductivity.
Supported features#
The CRYSTAL interface reads the unit cell from a CRYSTAL output file (lattice vectors, conventional atomic numbers, fractional atomic positions). For optimization outputs, the final geometry in the file is read.
If dielectric tensor and effective Born charges are available, they can be
read with phonopy-crystal-born script. The script creates a BORN file for
NAC.
The recommended strategy is to carry out a Gamma-point frequency calculation with INTENS and INTCPHF and use this as input to Phonopy. This produces all required quantities and also confirms that the structure optimization has converged to a true local minimum.
If ATOMSPIN keyword is present, magnetic moments are read from it. There is very little experience on using this feature, so be careful.
How to run#
The workflow for a CRYSTAL-Phonopy calculation is outlined here using the
Si example found in example/Si-CRYSTAL.
In this example, the CRYSTAL output file is crystal.o.
This is the default for the CRYSTAL interface, so, the -c crystal.o
parameter is not needed
Create supercells with --crystal option:
% phonopy-init --crystal -d --dim="4 4 4"
In this example, 4x4x4 supercells are created. For every supercell file, the interface creates a .d12 input file and an .ext structure file. The files
supercell.d12/.extcontain the perfect supercell. The filessupercell-xxx.d12/.ext(xxxare numbers) contain the supercells with displacements. Filephonopy_disp.yamlis also generated, containing information about the supercell and the displacements.In the case of the Si example, files
supercell-001.d12andsupercell-001.extwill be created.To make valid CRYSTAL input files, there are two possible options:
Manually: modify the generated supercell-xxx.d12 files by replacing the line
Insert basis sets and parameters herewith the basis set and computational parameters.Recommended option: before generating the supercells, include a file named
TEMPLATEin the current directory. This file should contain the basis sets and computational parameters for CRYSTAL (see the Si example). When phonopy finds this file, it automatically generates complete CRYSTAL input files in the step 1
Note that supercells with displacements must not be relaxed in the force calculations, because atomic forces induced by a small atomic displacement are what we need for phonon calculation. To get accurate forces, TOLDEE parameter should be 10 or higher. Phonopy includes this parameter and the necessary GRADCAL keyword automatically in the inputs.
Then, CRYSTAL supercell calculations are executed to obtain forces on atoms, e.g., as follows:
% runcry17 supercell-001.d12
To create
FORCE_SETSfile required by phonopy, the following command is executed:% phonopy-init --crystal -f supercell-001.o
Here
.ofiles are the CRYSTAL output files from the force calculations. All.ofiles corresponding to the generatedsupercell-xxx.d12files have to be given in the above command. To run this command,phonopy_disp.yamlhas to be located in the current directory because the information on atomic displacements stored inphonopy_disp.yamlare used to generateFORCE_SETS. See some more detail at CRYSTAL interface.Now, Phonopy post-prcessing commands can be run.
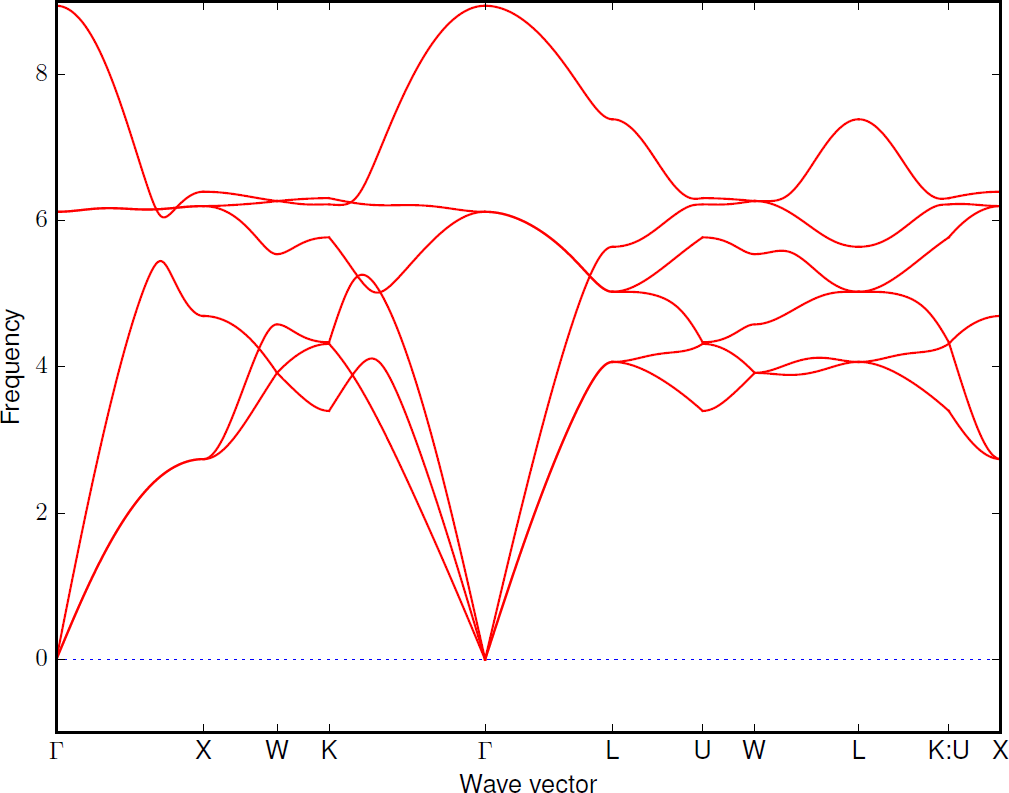
FORCE_SETSis automatically read in. Create phonon dispersion plot with:% phonopy -p -s band.conf _ _ __ | |__ ___ _ __ ___ _ __ _ _ | '_ \| '_ \ / _ \| '_ \ / _ \ | '_ \| | | | | |_) | | | | (_) | | | | (_) || |_) | |_| | | .__/|_| |_|\___/|_| |_|\___(_) .__/ \__, | |_| |_| |___/ 1.13.0 Python version 2.7.3 Spglib version 1.10.3 Calculator interface: crystal Band structure mode Settings: Supercell: [4 4 4] Spacegroup: Fd-3m (227) Computing force constants... max drift of force constants: 0.000000 (xx) 0.000000 (zz) Reciprocal space paths in reduced coordinates: [ 0.00 0.00 0.00] --> [ 0.50 0.00 0.50] [ 0.50 0.00 0.50] --> [ 0.50 0.25 0.75] [ 0.50 0.25 0.75] --> [ 0.37 0.38 0.75] [ 0.38 0.38 0.75] --> [ 0.00 0.00 0.00] [ 0.00 0.00 0.00] --> [ 0.50 0.50 0.50] [ 0.50 0.50 0.50] --> [ 0.63 0.25 0.63] [ 0.62 0.25 0.62] --> [ 0.50 0.25 0.75] [ 0.50 0.25 0.75] --> [ 0.50 0.50 0.50] [ 0.50 0.50 0.50] --> [ 0.37 0.37 0.75] [ 0.62 0.25 0.62] --> [ 0.50 -0.00 0.50] _ ___ _ __ __| | / _ \ '_ \ / _` | | __/ | | | (_| | \___|_| |_|\__,_|
For further settings and command options, see the general Phonopy documentation Setting tags and Command options, respectively, and for examples, see Examples.
Non-analytical term correction (Optional)#
The workflow for a CRYSTAL-Phonopy calculation with NAC is outlined here
using the NaCl example found in example/NaCl-CRYSTAL.
In this example, the CRYSTAL output file is crystal.o.
This is the default for the CRYSTAL interface, so, the -c crystal.o
parameter is not needed.
To activate non-analytical term correction, BORN (optional) is
required. This file contains the Born effective charges
and the dielectric tensor. They can be calculated with CRYSTAL.
The recommended strategy is to carry out a Gamma-point frequency calculation
with INTENS and INTCPHF. This produces all required quantities and also confirms that
the structure optimization has converged to a true local minimum.
(see the FREQCALC-INTENS-INTCPHF block in the beginning of crystal.o)
The workflow is very similar to the Si example below:
Create displaced supercells:
phonopy-init --crystal --dim="4 4 4" -d
Note that now the CRYSTAL interface automatically creates the
BORNfile. It should look like this:default 1.8126 0.0000 0.0000 0.0000 1.8126 0.0000 0.0000 0.0000 1.8126 1.0238 -0.0000 -0.0000 -0.0000 1.0238 0.0000 -0.0000 0.0000 1.0238 -1.0238 0.0000 0.0000 0.0000 -1.0238 0.0000 0.0000 0.0000 -1.0238
However, if you don’t want to run a FREQCALC-INTENS-INTCPHF calculation, but have the necessary data from some other source, you can create the
BORNfile manually following theBORNformat (BORN (optional)).Create
BORNfile withphonopy-crystal-bornscript:phonopy-crystal-born > BORN
By default,
phonopy-crystal-bornlooks for a file called crystal.o. File with some other name can be given as an argument to the script. TheBORNfile created by the script should look like this:default 1.8126 0.0000 0.0000 0.0000 1.8126 0.0000 0.0000 0.0000 1.8126 1.0238 0.0000 0.0000 0.0000 1.0238 0.0000 0.0000 0.0000 1.0238 -1.0238 -0.0000 -0.0000 -0.0000 -1.0238 0.0000 -0.0000 0.0000 -1.0238
If you don’t want to run a FREQCALC-INTENS-INTCPHF calculation, but have the necessary data from some other source, you can create the
BORNfile manually following theBORNformat (BORN (optional)).Run the supercell inputs with CRYSTAL as in the Si example above.
Collect forces:
phonopy-init --crystal -f supercell-*o
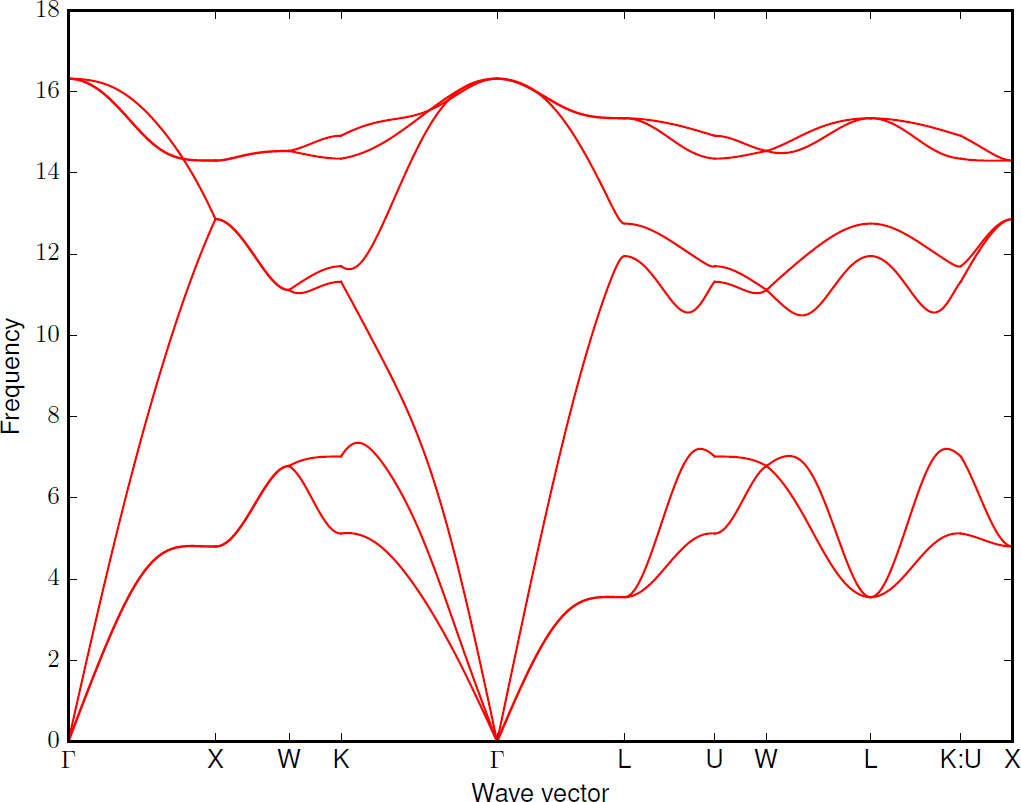
Calculate phonon dispersion data into band.yaml and save band.pdf, using non-analytical correction
--nac:phonopy -p -s --nac band.conf